
Why This Study Does Not Prove Omega-3s Are Harmful: A Critical Analysis of Liao et al. (2026)

A new study says omega-3 supplements accelerate cognitive decline.
The headline is alarming. The data are real. And the conclusion is almost certainly wrong — or at least, dangerously incomplete.
Here is why.
What the Study Actually Showed
Liao et al. analyzed 819 ADNI participants (273 omega-3 users, 546 matched non-users) over a median of 5 years. Link: https://doi.org/10.1016/j.tjpad.2026.100569
Omega-3 users showed faster decline on three cognitive measures (MMSE, ADAS-Cog13, CDR-SB). This decline was not mediated by amyloid, tau, or brain atrophy — but was partially mediated by declining FDG-PET signal, a marker of synaptic metabolic function.
The authors deserve credit for a thoughtful analysis. They checked for reverse causality (pre-supplementation trajectories were similar between groups), used propensity score matching, and were transparent about (it’s many) limitations.
But the study has critical gaps that prevent the conclusion its headline implies. And the most important gap is one the authors never considered.
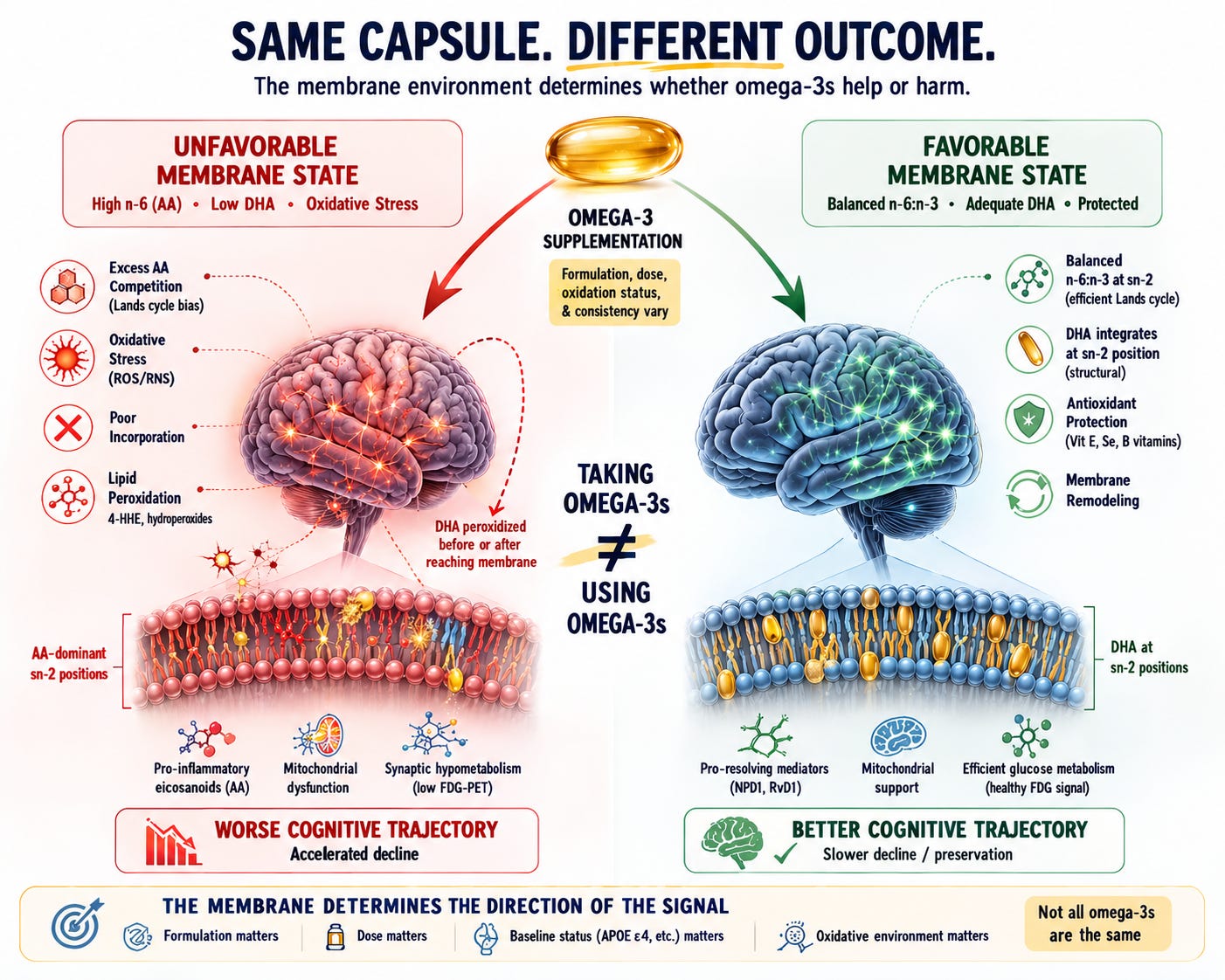
The Core Problem: The Membrane Was Never Measured
This is the central issue — not just with this study, but with nearly every omega-3 trial that has produced confusing results. It is also the principle that runs through this entire series here on The NeuroLipid Notebook:
The membrane decides.
DHA does not work by floating through the bloodstream. It works by incorporating into the sn-2 position of neuronal membrane phospholipids, where it serves as the substrate for specialized pro-resolving mediators like neuroprotectin D1, supports membrane fluidity, and modulates synaptic signaling. The entire biological rationale for DHA supplementation depends on this one event: successful incorporation into the membrane.
But incorporation is not guaranteed. It depends on what is already sitting at sn-2.
The dominant reacylation enzyme in the Lands cycle — LPCAT3 — has strong structural selectivity for arachidonoyl-CoA. Its crystal structure reveals a side pocket specifically shaped to hold arachidonic acid, and functional studies confirm that LPCAT3 specifically introduces AA at the sn-2 position of lysophosphatidylcholine. When LPCAT3 is silenced, AA content drops and EPA/DHA accretion increases — confirming that LPCAT3 activity actively excludes DHA from the sn-2 position of PC.
This means that in a membrane already dominated by AA at sn-2 — which is the expected state in individuals with a Standard American diet, high dietary omega-6 intake, chronic inflammatory signaling, or APOE4-driven lipid remodeling — incoming DHA faces a biochemical bottleneck. The reacylation machinery preferentially refills the vacated sn-2 position with AA, not DHA. Each cycle of cPLA₂α cleavage and LPCAT3-mediated refilling shifts the membrane further toward AA dominance.
AA and DHA also compete differently across phospholipid classes. AA is preferentially esterified into phosphatidylinositol (PI) with 8-fold higher efficiency than DHA, while DHA incorporates more evenly across PI and PC. In a membrane already saturated with AA at sn-2 positions, the available slots for DHA are reduced.
So what happens to DHA that arrives at the neuron but cannot incorporate?
It does not simply disappear. As the most unsaturated fatty acid in the brain — six double bonds — free or poorly incorporated DHA is exquisitely vulnerable to non-enzymatic peroxidation by reactive oxygen species. The products of DHA peroxidation include 4-hydroxyhexenal (4-HHE), an aldehyde that forms adducts with proteins, phospholipids, and nucleic acids. A mouse study showed that a DHA-enriched diet substantially increased 4-HHE specifically in the hippocampus — the exact brain region most vulnerable in Alzheimer’s disease.
These peroxidation products directly impair mitochondrial function. They collapse membrane potential, inhibit oxidative phosphorylation, and paradoxically generate more reactive oxygen species — creating a self-reinforcing cycle of oxidative damage.
This would produce exactly the pattern the Liao et al. study found: declining FDG signal (glucose hypometabolism reflecting mitochondrial dysfunction) without corresponding changes in amyloid, tau, or brain volume.
Normally, DHA triggers its own protection. When DHA successfully incorporates into hippocampal membranes, it upregulates GPx4 — the enzyme that reduces phospholipid hydroperoxides in situ — as a self-protective mechanism. But this upregulation is significantly attenuated in APP/PS1 transgenic mice, a model of familial Alzheimer’s disease. This means that in an AD-vulnerable brain, the self-protection mechanism that normally accompanies DHA incorporation is already compromised.
Here is my critique: the Liao et al. study did not measure membrane fatty acid composition at any point. It did not know whether participants’ neuronal membranes were AA/AdA-dominant or DHA-replete. It did not know whether the supplemental DHA was incorporating successfully or peroxidizing. It did not know whether the antioxidant systems required to protect newly incorporated DHA were functional.
The variables that determine whether DHA helps or harms was never measured.
And there is a striking counterpoint. A cross-sectional study in 320 cognitively unimpaired participants at increased AD risk found the opposite of the Liao et al. result: blood omega-3 levels were directly associated with preserved FDG uptake in AD-vulnerable brain regions, with stronger associations in APOE4 carriers and homozygotes. This suggests that endogenous omega-3 status — reflecting successful incorporation and utilization — supports glucose metabolism. The Liao et al. finding may reflect something specific to the supplements (oxidation, formulation, dose, membrane context) rather than to omega-3 biology itself.
A separate ADNI analysis using erythrocyte omega-3 index found that low omega-3 was associated with greater amyloid accumulation and greater memory decline — and among APOE4 carriers specifically, low omega-3 was associated with higher tau accumulation.
The same molecule. Opposite outcomes. The difference is not the molecule — it is the membrane environment receiving it.
This is not a theoretical concern. It is the central uncontrolled variable in this study and in most omega-3 trials. Taking DHA is not the same as using DHA.
The membrane decides.
Problem 1: They Did Not Know What People Were Actually Taking — And Much of It May Not Have Been DHA
The study classified participants as “omega-3 users” based on self-reported supplement use. The primary formulation was commercial fish oil. But the study could not determine the dose, the formulation (ethyl ester vs. triglyceride vs. phospholipid), the oxidation status of the product, or whether participants actually took it consistently.
This dose blindness is not a minor issue. A JAMA Cardiology analysis of 255 fish oil supplements from 16 leading U.S. brands found enormous variability: median EPA was 340 mg/day, median DHA was 270 mg/day, and median total EPA+DHA was only 600 mg/day. Only 9.4% of products contained ≥2 g/day EPA+DHA. A separate analysis of 47 commercial supplements found that over 70% did not contain the stated label amount of EPA or DHA.
Crucially, the typical commercial fish oil supplement delivers EPA and DHA in approximately a 1.3:1 ratio (median 340 mg EPA vs. 270 mg DHA), meaning participants were likely getting more EPA than DHA. This matters profoundly for the brain. DHA — not EPA — is the dominant omega-3 in neuronal membranes, comprising 30–40% of phospholipid fatty acids in the cerebral cortex and synaptic membranes. EPA, by contrast, is present in brain tissue at only trace levels (~0.1–0.3% of total fatty acids even after supplementation) and serves primarily as a circulating anti-inflammatory precursor. A 2024 animal study in 3xTg-AD mice confirmed that even after high-dose EPA supplementation, brain EPA reached only 0.29% of total fatty acids, while DHA drove cortical phospholipid enrichment.
The cognitive benefit literature consistently points to DHA as the critical molecule for brain outcomes. A meta-analysis of 48 cohort studies found that each 0.1 g/day increment of DHA intake was associated with an 8–10% lower risk of cognitive decline — and erythrocyte membrane DHA (not EPA) was the blood marker most consistently associated with reduced dementia risk. The VITACOG trial found that DHA specifically (not EPA) drove the interaction between omega-3 status and B vitamin efficacy on brain atrophy.
If the ADNI omega-3 users were taking standard commercial fish oil at typical doses — which the data strongly suggest — they were getting roughly 270 mg/day of DHA. This is well below the 900 mg/day dose that showed cognitive benefit in the one positive RCT, and far below the doses used in most clinical trials.
The study also lumped together fish oil, krill oil, and flaxseed oil under the single umbrella of “omega-3 supplementation.” This introduces a biologically heterogeneous exposure that cannot be interpreted as a single intervention, because flaxseed oil contains no DHA or EPA whatsoever. It contains alpha-linolenic acid (ALA), an 18-carbon plant-derived omega-3 that must undergo a long enzymatic conversion chain before it becomes DHA.
The conversion rate of ALA to DHA in humans is extremely poor: approximately 0–4% in men and modestly higher in premenopausal women due to estrogen’s upregulation of desaturase enzymes. The American College of Cardiology has noted that ALA conversion to EPA is only about 10%, and to DHA approximately 1%. Multiple reviews have concluded that “conversion of both ALA and SDA to DHA is limited in most humans.” The conversion is further impaired by high dietary linoleic acid (LA) intake — the standard Western diet — because LA and ALA compete for the same desaturase enzymes. Genetic variation in the FADS1-2-3 gene cluster adds another layer of individual variability. (And for more on FADS genotype: read this:
https://neurolipidnotebook.substack.com/p/the-omega-6-paradox
This means that a participant taking flaxseed oil was receiving a fundamentally different biological intervention than one taking fish oil. The flaxseed oil user was getting almost no preformed DHA for neuronal membrane incorporation. Grouping these participants together with fish oil users — and then concluding that “omega-3 supplementation” caused cognitive decline — is like grouping aspirin and acetaminophen users together and concluding that “pain relievers” cause liver damage.
Finally, the study could not determine the oxidation status of the supplements. Multiple independent analyses have found that commercial fish oil supplements frequently exceed international oxidation limits — with 24–83% of products exceeding recommended peroxide values depending on the market. One U.S. analysis found that omega-3 fatty acids isolated from dietary supplements were too oxidized to inhibit LDL oxidation, while non-oxidized forms inhibited it by >95%.
The authors themselves acknowledge this: “fish oil, the primary supplement type, is particularly susceptible to oxidation... the observed harmful association may not generalize to other omega-3 formulations with greater oxidative stability.”
In other words: this study may be measuring the combined effect of low-dose, EPA-dominant, potentially oxidized fish oil and biologically inert flaxseed oil — not the effect of DHA on the brain.
Problem 2: They Did Not Measure the Nutrients That Determine Whether DHA Helps or Harms
This omission compounds the membrane problem. Even if DHA reaches the neuron, its safe incorporation and protection depend on co-nutrients the study never measured:
- Vitamin E status (the primary chain-breaking antioxidant protecting DHA in membranes)
- Selenium status (required for GPx4, the enzyme that reduces phospholipid hydroperoxides in situ — the same enzyme DHA normally upregulates for self-protection)
- B vitamin status or homocysteine levels (which determine whether omega-3 produces cognitive benefit)
- Vitamin D status (which interacts with omega-3 in cognitive outcomes)
Why does this matter? Because we have strong randomized trial evidence that these co-nutrients determine the direction of omega-3’s effect on the brain.
The VITACOG trial — a randomized, placebo-controlled study in 168 people with mild cognitive impairment — found that B vitamin treatment slowed brain atrophy by 40%. But only in participants who also had high omega-3 levels. In those with low omega-3, B vitamins did nothing. And the reverse was also true: omega-3 levels only predicted slower atrophy in the B vitamin group, not in the placebo group (interaction p = 0.024).
This bidirectional interaction has been replicated in at least four independent trials:
- The OmegAD trial found that omega-3 supplementation improved cognition in Alzheimer’s patients — but only in those with adequate B vitamin status (homocysteine below 11.7 μmol/L). In those with poor B vitamin status, omega-3 had no benefit.
- The FACIT trial (n=791) found that folic acid improved cognition — but the effect was dependent on baseline omega-3 status.
- The B-Proof trial (n=191) found that B vitamin efficacy on cognition was linked specifically to DHA status.
- A meta-analysis of 14 RCTs (n=4,913) found significant benefit of multi-nutrient formulas combining omega-3 + B vitamins on global cognition (effect size 0.23, p = 0.002) and episodic memory (effect size 0.32, p = 0.001).
A 2025 metabolomics analysis of the VITACOG trial revealed that B vitamins induced broad metabolic reprogramming — lowering quinolinic acid, α-ketoglutarate, and glutamate — affecting TCA cycle and glutamine-glutamate cycling critical for brain energy homeostasis. This is directly relevant to the FDG hypometabolism finding in the Liao et al. study.
The Liao et al. study did not measure any of these variables. It is entirely possible — even likely — that the omega-3 users who declined fastest were those with poor B vitamin status, low vitamin E, or low selenium, in whom supplemental DHA could not be safely incorporated into neuronal membranes and instead underwent peroxidation — feeding the exact mitochondrial damage pathway described above.
Problem 3: The Broader Evidence Strongly Favors Omega-3
This single observational study must be weighed against a much larger body of evidence:
- A meta-analysis of 48 prospective cohort studies (103,651 participants) found that dietary omega-3 intake reduced dementia risk by approximately 20%, with each 0.1 g/day increment of DHA associated with an 8–10% lower risk of cognitive decline.
- The UK Biobank (440,750 participants, 7,768 dementia cases) found fish oil supplementation associated with a 7% lower dementia risk, mediated by plasma omega-3 levels.
- A dose-response meta-analysis of 21 cohort studies (181,580 participants) found that each additional serving of fish per week was associated with lower risks of dementia and Alzheimer’s disease.
- In the same ADNI cohort that Liao et al. used, a prior analysis found that long-term omega-3 supplement users had a 64% reduced risk of Alzheimer’s disease.
- The WHIMS-MRI study (1,111 postmenopausal women) found that higher RBC EPA+DHA was correlated with larger total brain volume and hippocampal volume 8 years later.
One observational study showing harm does not overturn this evidence base. It raises questions — important ones — but it does not “prove” omega-3s are harmful.
Problem 4: Massive Missing Data
The study’s baseline characteristics table reveals alarming levels of missing data. Education was missing for 58% of participants. Comorbidity data (stroke, anxiety, depression, insomnia, Parkinson’s) were missing for 60–88% of participants. With this degree of missingness, unmeasured confounding is not just possible — it is likely.
The study also did not stratify by APOE4 status in its primary analysis, despite including APOE4 as a covariate. The authors acknowledge they lacked power for subgroup analyses. This is a significant limitation given the APOE4-specific effects on DHA transport and membrane metabolism. The Rouch et al. ADNI analysis found that the omega-3–tau association was specific to APOE4 carriers — suggesting that APOE4 status may be a critical effect modifier that the Liao et al. study could not adequately address. Notably, Rouch et al. used the same ADNI cohort as Liao et al. — but measured erythrocyte omega-3 index rather than self-reported supplement use — and found that low tissue omega-3 was associated with greater amyloid accumulation, faster memory decline, and higher tau burden specifically among APOE4 carriers: the opposite direction of effect from the same dataset.
What This Study Actually Supports
Ironically, this study’s findings are most consistent not with the conclusion that omega-3s are harmful, but with the principle that runs through this entire series: the membrane decides.
The FDG hypometabolism finding — synaptic metabolic failure without amyloid, tau, or atrophy changes — is precisely what you would predict if supplemental DHA were peroxidizing rather than incorporating. And DHA peroxidation is precisely what you would predict if:
- The membrane was already AA-dominant at sn-2, blocking DHA incorporation via LPCAT3 selectivity
- The supplement was already oxidized before ingestion
- The antioxidant systems (GPx4, vitamin E) were insufficient to protect newly incorporated DHA
- The self-protective GPx4 upregulation that DHA normally triggers was attenuated (as it is in AD models)
All four of these conditions are plausible — even likely — in the ADNI cohort, which included participants with MCI and AD, taking uncharacterized commercial supplements, with unmeasured antioxidant and B vitamin status, and unmeasured membrane composition.
DHA is not unconditionally beneficial. It requires:
- A membrane that can receive it — meaning sn-2 positions not already locked by AA through LPCAT3-mediated reacylation bias
- Adequate antioxidant protection — vitamin E and selenium to prevent its peroxidation in membranes (Consult with your own healthcare provider to determine optimal doses for you, this is not to be taken as individualized medical advice)
- Adequate B vitamin status to support its neuroprotective effects
- A supplement that is not already oxidized when swallowed
- The supplement to actually contain meaningful doses of preformed DHA — not ALA from flaxseed oil or EPA-dominant fish oil at subtherapeutic doses
When these conditions are met, the evidence consistently shows benefit. When they are not met — when low-dose, EPA-dominant, potentially oxidized fish oil or biologically inert flaxseed oil enters a brain whose membranes are already AA-saturated and whose antioxidant defenses are depleted — the same molecule that should protect neurons may damage their mitochondria instead.
Taking omega-3s ≠ using omega-3s.
The membrane decides.
This study, inadvertently, may be the strongest argument yet for getting that preparation right.
This article is part of a series. If you’re starting here, I have several other articles that compliment this one in different ways. Browse my home-page or consider reading this next:

Please keep in mind that while I am a Nurse Practitioner by training, this article and my entire NeuroLipid Notebook archive are my personal intellectual contributions and positions and are not to be taken as individual medical advice or proven theories.
If you liked this post, you’ll LOVE The NeuroLipid Notebook. Subscribe and say hello!
References
Liao ZB, Hu ZC, Zeng GH, et al. The association between omega-3 supplementation and cognitive decline in older adults. J Prev Alzheimers Dis. 2026;100569. doi:10.1016/j.tjpad.2026.100569
Zhang Q, Yao D, Rao B, et al. The structural basis for the phospholipid remodeling by lysophosphatidylcholine acyltransferase 3. Nat Commun. 2021;12:6869.
Sun GY, Simonyi A, Fritsche KL, et al. DHA supplementation alters phospholipid species and lipid peroxidation products in adult mouse brain, heart, and plasma. Neuromolecular Med. 2021;23:118-129.
Bradley MA, Markesbery WR, Lovell MA. Elevated 4-hydroxyhexenal in Alzheimer’s disease (AD) progression. Neurobiol Aging. 2012;33:1034-1044.
Baker EJ, Miles EA, Burdge GC, Yaqoob P, Calder PC. Metabolism and functional effects of plant-derived omega-3 fatty acids in humans. Prog Lipid Res. 2016;64:30-56.
Mason RP, Sherratt SCR. Omega-3 fatty acid fish oil dietary supplements contain saturated fats and oxidized lipids that may interfere with their intended biological benefits. Biochem Biophys Res Commun. 2017;483:425-429.
Jernerén F, Elshorbagy AK, Oulhaj A, Smith SM, Refsum H, Smith AD. Brain atrophy in cognitively impaired elderly: the importance of long-chain ω-3 fatty acids and B vitamin status in a randomized controlled trial. Am J Clin Nutr. 2015;102:215-221.
Fairbairn P, Tsofliou F, Johnson A, et al. The effects of multi-nutrient formulas containing a combination of n-3 PUFA and B vitamins on cognition in the older adult: a systematic review and meta-analysis. Br J Nutr. 2023;129:428-441.
Klein EA, Thompson IM, Tangen CM, et al. Vitamin E and the risk of prostate cancer: the Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA. 2011;306:1549-1556.
Wei BZ, Li L, Dong CW, Tan CC, Xu W. The relationship of omega-3 fatty acids with dementia and cognitive decline: evidence from prospective cohort studies of supplementation, dietary intake, and blood markers. Am J Clin Nutr. 2023;117:1096-1109.
Lázaro I, Grau-Rivera O, Suárez-Calvet M, et al. Omega-3 blood biomarkers relate to brain glucose uptake in individuals at risk of Alzheimer’s disease dementia. Alzheimers Dement (Amst). 2024;16:e12596.
Rouch L, Virecoulon Giudici K, Cantet C, et al. Associations of erythrocyte omega-3 fatty acids with cognition, brain imaging and biomarkers in the Alzheimer’s Disease Neuroimaging Initiative: cross-sectional and longitudinal retrospective analyses. Am J Clin Nutr. 2022;116:1492-1506.