

Why Ergothioneine May Be the Missing Piece in APOE4 Brain Defense
Ergothioneine may act as an upstream defense against oxidative stress and ferroptosis in the brain.

Where This Article Fits in the NeuroLipid Notebook Series
The NeuroLipid Notebook maps how APOE4 rewires the brain’s lipid metabolism — from the initial fork between inflammation and resolution (The Vortex), through the supply failure that starves the brain of DHA (The Drain), to the self-reinforcing loop that replaces DHA with ferroptotic substrates (The Engine), and finally to the three death pathways that fire when every rescue mechanism has been locked (The Kill Switches and The Narrowing Margin).
This article steps outside the membrane to ask a different question: is there a defense molecule the brain actively imports from the diet — one that operates upstream of the entire ferroptotic cascade — that APOE4 carriers may be burning through faster than they can replace?
No prior articles are required to follow this one. But readers familiar with the series will recognize where ergothioneine’s iron chelation, arachidonic acid peroxidation inhibition, and NRF2 activation map onto the framework — and why the absence of APOE4-stratified data in the ergothioneine literature mirrors the exact problem the series was built to expose.
New here? Start with The Vortex for the full framework, or read on — this article stands on its own.
In the last article, we explored how a landmark APOE-stratified proteomics study revealed that 64–78% of Alzheimer’s-associated proteins had been invisible to every prior study — hidden by the practice of pooling APOE genotypes together. Among the metabolomic findings was a quietly devastating detail: ergothioneine — a diet-derived antioxidant with its own dedicated brain transporter — was consistently depleted in AD patients across APOE genotypes. I had to really dig into the supplemental data to find it for you all, btw!
BTW, you can catch up on that article here if you missed it:

That finding deserves its own article. Because ergothioneine may be the molecule that connects the dots between mushroom consumption and dementia risk, between iron accumulation and neuronal death, and between what we know about APOE4 biology and what we can actually do about it — now, safely, and without a prescription.
What Is Ergothioneine, and Why Does the Brain Want It So Badly?
Ergothioneine is a sulfur-containing amino acid produced exclusively by certain fungi and bacteria. Humans cannot synthesize it. We get it almost entirely from mushrooms, with smaller amounts from beans, oats, and organ meats.
Here is what makes ergothioneine unusual: despite being a molecule we cannot make, the human body has evolved a dedicated transporter — OCTN1, encoded by the gene SLC22A4 — whose primary function appears to be absorbing and distributing ergothioneine to tissues throughout the body, including the brain. OCTN1 is expressed in neurons, neural stem cells, and microglia. Mice engineered to lack OCTN1 have zero ergothioneine in their brains. The transporter is so specific that one research group renamed it the “ergothioneine transporter” (ETT), arguing that ergothioneine is its primary physiological substrate.
The existence of a dedicated transporter for a molecule we cannot synthesize is a strong evolutionary signal. It suggests that ergothioneine provides something the body cannot get from its own biochemistry — something important enough that natural selection built specialized infrastructure to acquire it from the environment.
Paul and Snyder (2010) demonstrated what that something might be: cells depleted of the ergothioneine transporter showed increased mitochondrial DNA damage, protein oxidation, and lipid peroxidation. Ergothioneine concentrates in mitochondria, where it appears to specifically protect mitochondrial components from oxidative damage. The authors described ergothioneine as “a physiologic cytoprotectant,” and subsequent reviews have proposed that it may even warrant classification as a new vitamin — a diet-derived molecule essential for cellular defense that the body cannot produce on its own.
The Epidemiological Signal: Mushrooms, Dementia, and a Dose-Response Gradient
Before diving into the molecular mechanisms, the population-level data deserve attention — because they are remarkably consistent across three countries and five independent cohorts.
The Ohsaki Cohort (2017) followed 13,230 elderly Japanese individuals for 5.7 years. Those who consumed mushrooms three or more times per week had a 19% lower risk of incident dementia compared to those who consumed mushrooms less than once per week (HR 0.81, 95% CI 0.69–0.95), after adjusting for age, sex, BMI, smoking, alcohol, exercise, and dietary factors.
The CIRCS Study (2024) followed approximately 3,750 Japanese adults for 16 years and found that mushroom intake above 15 g/day was associated with a 44% lower risk of disabling dementia (HR 0.56, 95% CI 0.42–0.75) — but only in women. In men, no association was observed.
That sex-specific finding should stop us in our tracks. It echoes exactly what Li and Wretlind et al. (2026) found in the lipidomic data we discussed in the last article (The Biggest Alzheimer’s Study You Haven’t Heard About): lipid-AD associations were almost entirely female-driven. And it raises a question we will return to in future articles: why would a dietary antioxidant protect women’s brains more than men’s? The answer may have everything to do with what happens to brain membrane defenses when estrogen withdraws at menopause — but that is a story for another day.
The Singapore DaHA study (2019) found that consuming more than two portions of mushrooms per week was associated with 57% lower odds of MCI (OR 0.43, 95% CI 0.23–0.78). A US NHANES analysis (Ba et al. 2022) found that higher mushroom intake was associated with better cognitive performance on processing speed and word learning tests in adults over 60.
The Hisayama Study (Meng et al. 2025) provides the strongest evidence to date — and it eliminates the most significant limitation of every study listed above. Unlike the dietary studies, which relied on participants remembering what they ate, the Hisayama Study measured serum ergothioneine directly using LC-MS/MS. Among 1,344 community-dwelling Japanese adults aged 65 and over, followed for a median of 11.2 years, 273 developed dementia (201 AD, 72 non-AD). The risk of all-cause dementia, AD, and non-AD dementia decreased progressively across increasing quartiles of serum ergothioneine — a dose-response gradient, not just a threshold effect — and the trend remained significant after adjusting for cardiovascular risk factors, lifestyle, and dietary factors including daily vegetable intake. Most critically, when the investigators stratified by vegetable consumption, higher serum ergothioneine was associated with lower dementia risk regardless of how many vegetables participants ate. This rules out the most obvious confounding explanation — that ergothioneine is simply a marker of a healthy diet. The signal appears to be specific to ergothioneine itself.
These are observational studies with all the usual caveats — confounding by healthy lifestyle, reverse causation, and in the dietary studies, recall bias. But the consistency across three countries, five independent cohorts, and different cognitive endpoints is notable — and the Hisayama Study’s biomarker-based design and vegetable-stratified analysis address the two most common objections. The biological plausibility is strong, because we know exactly how ergothioneine gets into the brain and what it does when it arrives.
The Biomarker Evidence: Ergothioneine Declines With Cognitive Decline
The observational data become more compelling when paired with direct biomarker measurements.
Wu et al. (2021) measured plasma ergothioneine in 496 participants across the cognitive spectrum — cognitively normal, cognitive impairment no dementia (CIND), and dementia. Ergothioneine was lowest in dementia, intermediate in CIND, and highest in controls, with a stepwise decline tracking disease severity (P 0.001). Lower ergothioneine was independently associated with brain atrophy, reduced cortical thickness, hippocampal volume loss, and white matter hyperintensities — after adjusting for demographic and vascular risk factors.
Wu et al. (2022) then followed 470 elderly subjects longitudinally for up to five years. Lower baseline plasma ergothioneine predicted faster rates of decline in memory, executive function, attention, visuomotor speed, and language — but only in non-demented individuals. This is a critical detail: the predictive power was concentrated in the window before irreversible damage, suggesting that ergothioneine depletion is an early event, not a late consequence.
Cheah et al. (2016) — the original observation — found that whole blood ergothioneine levels decline significantly beyond age 60. The timing is not coincidental. Age 60 is the same decade when multiple brain defense systems begin to falter: the transporter that delivers DHA across the blood-brain barrier declines, the enzyme that strips DHA from membranes (cPLA₂) increases, and in women, the loss of estrogen removes an entire layer of antioxidant protection. If APOE4 carriers are consuming ergothioneine faster due to elevated oxidative burden, their functional depletion may begin even earlier — potentially in the late 40s or 50s.
Most recently, Eslick et al. (2026) found that among cognitively normal people who already have brain amyloid (Aβ+ on PET), higher plasma ergothioneine correlates with a more favorable Aβ42/40 ratio. This suggests that ergothioneine may be modifying amyloid processing even in the presymptomatic amyloid-positive phase — precisely the stage at which APOE4 carriers spend years before clinical symptoms emerge.
The First Clinical Trial — and the Larger One Coming
Yau et al. (2024) conducted the first randomized, double-blind, placebo-controlled trial of ergothioneine in MCI — 19 subjects, 25 mg three times weekly for one year. Despite the tiny sample, the ergothioneine group showed improved learning ability on the Rey Auditory Verbal Learning Test and — critically — stabilized neurofilament light chain (NfL) levels, while the placebo group showed a significant NfL increase. NfL is a marker of neuronal damage; its stabilization suggests that ergothioneine may be slowing neurodegeneration at the axonal level. No safety concerns were identified.
Nineteen subjects is far too few to draw clinical conclusions. But NfL stabilization in a one-year trial is a meaningful biological signal — it means something is changing at the level of neuronal integrity, not just subjective cognition.
Now a much larger trial is coming — and it is the first to explicitly recruit APOE4 carriers. Shan et al. (2025) published the protocol for a 24-month RCT of approximately 600 participants aged 45–74, selected specifically for being at elevated AD risk: family history of dementia, APOE ε4 carrier status, or subjective cognitive decline. The intervention is daily Pleurotus citrinopileatus (golden oyster mushroom) powder containing 7.0 mg/g ergothioneine by dry weight, with cognitive outcomes measured at baseline, 12 months, and 24 months using MMSE, Rey Auditory Verbal Learning Test, Symbol Digit Modality Test, and Clinical Dementia Rating. The trial also includes biological markers — though the specific panel has not been detailed in the published protocol.
This trial matters for three reasons. First, it is 30 times larger than the Yau et al. pilot. Second, it is 24 months rather than 12 — long enough to detect meaningful cognitive trajectories. Third, and most importantly, it is the first ergothioneine-relevant trial to include APOE4 carrier status as an enrollment criterion. If the investigators pre-specify APOE-stratified analyses, this trial could answer the single most important question in the ergothioneine-AD field: does the benefit differ by genotype?
The trial is also notable for what it uses as the intervention: whole mushroom powder rather than purified ergothioneine. This means participants receive the full complement of mushroom bioactives — beta-glucans, glutathione, vitamin D precursors, and other compounds — alongside ergothioneine. If the trial is positive, it will not be possible to attribute the benefit to ergothioneine alone. But from a practical standpoint, this may not matter: if the goal is to identify a safe, accessible, food-based intervention for at-risk individuals, whole mushroom powder is more translatable than a purified compound.
What Ergothioneine Actually Does at the Molecular Level — and Why It Matters for APOE4
This is where the story becomes specific to APOE4 carriers.
One of the defining features of APOE4 biology is elevated brain iron. Ayton et al. (2015) showed in a large longitudinal study that CSF ferritin — a marker of brain iron stores — is elevated in proportion to the number of APOE4 alleles a person carries, and that higher CSF ferritin predicted faster cognitive decline and MCI-to-AD conversion over seven years. Iron is not merely a bystander in neurodegeneration. It is the catalyst. Free iron drives Fenton chemistry — the reaction that converts relatively stable lipid hydroperoxides into highly reactive radicals that tear through cell membranes. This is the core chemistry of ferroptosis, the iron-dependent form of cell death that is increasingly recognized as central to Alzheimer’s pathology.
Ergothioneine hits this system at multiple points:
First, iron chelation. Ergothioneine is a potent chelator of iron and copper ions, inhibiting iron-dependent generation of hydroxyl radicals. Salama and Omar (2021) demonstrated that ergothioneine attenuates iron overload-induced hepatocellular injury in rats by modulating NF-κB, MAPK, and PI3K/AKT signaling — and notably, it curbed hepatic iron load, possibly through iron chelation. For an APOE4 carrier with elevated brain iron, ergothioneine would reduce the catalytic iron available to drive the membrane-destroying Fenton chemistry.
Second, direct inhibition of arachidonic acid peroxidation. Akanmu et al. (1991) — the foundational antioxidant characterization — showed that ergothioneine inhibits arachidonic acid peroxidation promoted by myoglobin/H₂O₂ mixtures. This is directly relevant: arachidonic acid peroxidation within membrane phospholipids is the proximate event in ferroptotic cell death. APOE4 carriers have overactive cPLA₂, the enzyme that liberates arachidonic acid from membranes — meaning they have more of the exact substrate that ergothioneine protects against.
Third, mitochondrial protection. Paul and Snyder (2010) showed that ergothioneine concentrates in mitochondria and specifically protects mitochondrial DNA from oxidative damage. The Li/Cruchaga APOE-stratified study found that mitochondrial dysfunction was the dominant APOE-independent pathway in AD. Ergothioneine’s mitochondrial localization positions it at the origin point of the oxidative stress that drives neurodegeneration.
Fourth, NRF2 pathway activation. Li et al. (2025) showed that ergothioneine synergistically enhanced neuroprotection in APP/PS1 mice through Keap1/NRF2-mediated mechanisms. NRF2 is the master transcription factor for antioxidant gene expression, including GPX4 — the enzyme that directly neutralizes the lipid peroxides generated during ferroptosis. Selenium provides the raw material for GPX4 synthesis; ergothioneine may help ensure the gene is turned on in the first place. For more on ferroptosis, read:

Whitmore et al. (2022) provided the most compelling preclinical evidence: longitudinal ergothioneine consumption — started before heavy plaque burden — reduced amyloid plaques and restored glucose metabolism in 5xFAD mice. This is a prevention paradigm, not a rescue paradigm. The benefit came from sustained early exposure, not late intervention. A second independent mouse study (Li et al. 2025) confirmed the cognitive benefit in APP/PS1 mice over six months, adding evidence that ergothioneine also suppresses microglial overactivation and reshapes gut microbiota — suggesting multiple converging mechanisms beyond direct antioxidant activity.
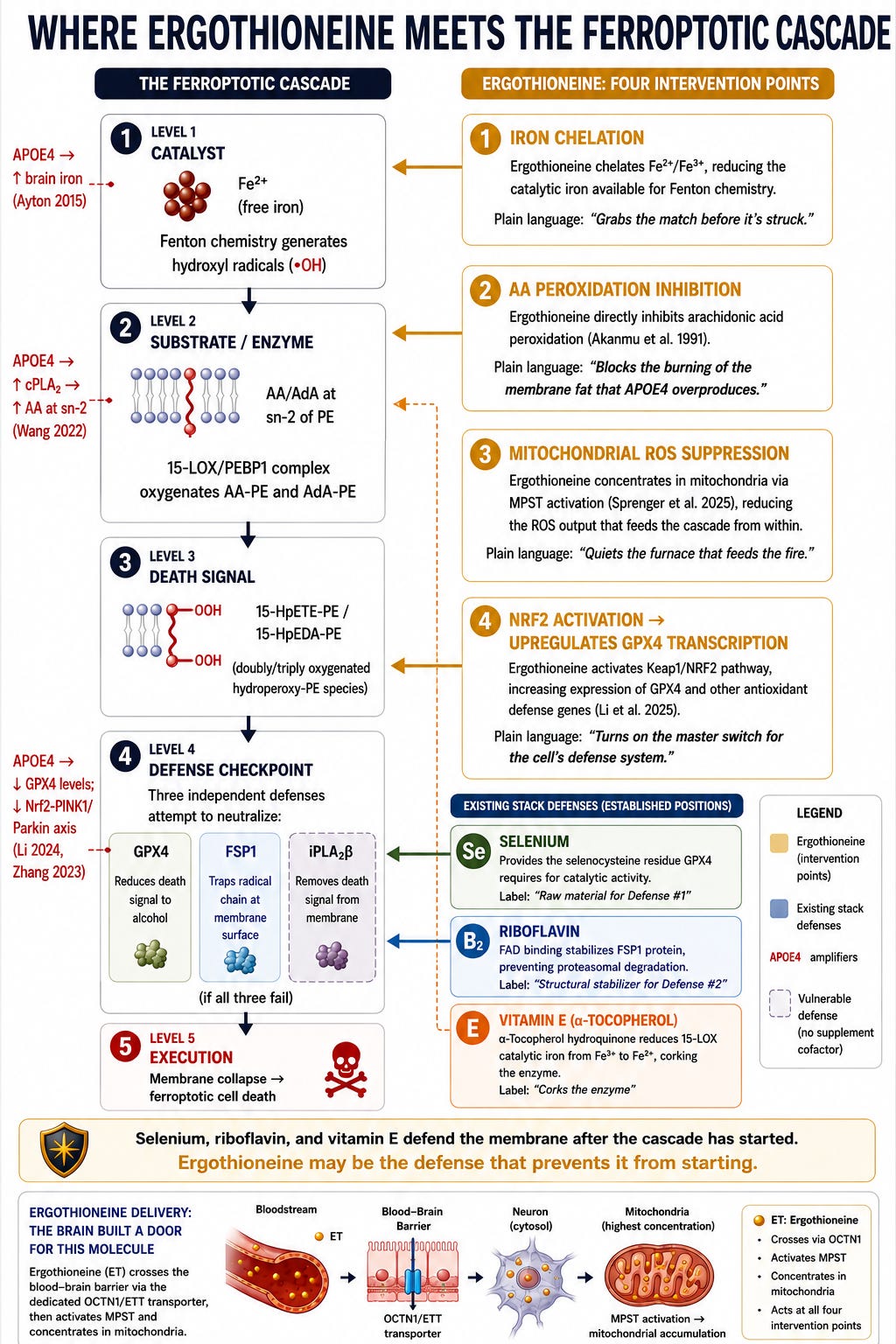
The Critical Gap: No One Has Asked the APOE4 Question
Here is the most important finding from the literature review: no published study has examined whether ergothioneine levels differ by APOE genotype, or whether the ergothioneine-cognition association is modified by APOE4 carrier status.
The Wu et al. studies adjusted for vascular risk factors but did not report APOE genotype. The Cheah et al. study did not genotype participants. The Yau et al. trial did not stratify by APOE. The mushroom-dementia epidemiological studies did not examine APOE interaction. Even the Shan et al. trial, which recruits APOE4 carriers, has not yet indicated whether APOE-stratified analyses are pre-specified.
This is a remarkable gap. APOE4 is the strongest genetic risk factor for AD, carried by approximately 25% of the population and approximately 60% of AD patients. If ergothioneine depletion is amplified in APOE4 carriers — because their elevated iron, overactive cPLA₂, and weakened antioxidant defenses burn through it faster — then the entire ergothioneine-AD literature may be underestimating the effect in the population that needs it most, while diluting it with a population that may have adequate endogenous defense.
This is exactly the same problem that the Li/Cruchaga APOE-stratified study exposed for proteomics: when you pool genotypes together, you miss genotype-specific signals. The ergothioneine field has not yet asked the APOE-stratified question directly.
This is exactly the same problem that the Li/Cruchaga APOE-stratified study exposed for proteomics: when you pool genotypes together, you miss genotype-specific signals. The ergothioneine field has not yet asked the APOE-stratified question directly.
The Li/Cruchaga dataset offers a preliminary answer. In their metabolomics analysis, ergothioneine was among the downregulated metabolites in AD cases. Their scatter plot of metabolite effect sizes across APOE genotypes showed that approximately 82% of differentially expressed metabolites had consistent direction in both ε3/ε3 and ε3/ε4 — and ergothioneine appears to fall in this consistent-down category. Separately, the study’s signaling network analysis (Figure S13B) identified ROS signaling and lipid signaling as consistently suppressed pathways in AD across both genotypes — the same pathways that ergothioneine’s established mechanisms (iron chelation, AA peroxidation inhibition, NRF2 activation) would be predicted to support — and their drug repositioning analysis independently identified iron chelation as a therapeutic strategy for the upregulated protein networks. Whether ergothioneine was specifically flagged in the study’s druggable target analysis requires clarification from the authors.
This suggests that ergothioneine depletion tracks AD pathology itself, regardless of APOE genotype. But the APOE4-specific question remains open: APOE4 carriers enter the disease process with higher brain iron, more active cPLA₂, and weaker antioxidant defenses. Even if the depletion rate is similar across genotypes, APOE4 carriers likely reach the critical threshold sooner — arriving at the same destination faster.
Three scenarios remain possible, and distinguishing between them requires genotype-stratified analysis of the metabolomics data with individual-level effect sizes:
If the depletion is similar across genotypes, ergothioneine is being consumed by the oxidative burden of AD itself — making it relevant for all AD patients.
If the depletion is worse in ε3/ε4, ergothioneine is being consumed faster in APOE4 carriers — strengthening the case for early, proactive supplementation in this population.
If the depletion is worse in ε3/ε3, it may represent a compensatory pathway more relevant to non-APOE4 AD — one of the mechanisms through which the 40% of AD patients who carry no APOE4 allele develop the disease through different biological routes.
I’ve reached out to one of the authors and will update this post if I hear back.
A Striking Omission in the Ferroptosis Literature
Perhaps the most surprising finding from this review is what is absent: no study has directly tested ergothioneine as a ferroptosis inhibitor using the standard ferroptosis assays. Ergothioneine does not appear in any of the major ferroptosis inhibitor reviews.
This is a striking omission given its iron chelation properties, its hydroxyl radical scavenging, its inhibition of arachidonic acid peroxidation, and its NRF2 activation — all of which are established anti-ferroptotic mechanisms. The experiment needed is straightforward: test ergothioneine against standard ferroptosis inducers in neuronal cell lines, with and without GPX4 inhibition, to determine whether it provides GPX4-independent ferroptosis protection.
If it does — as its iron chelation and arachidonic acid peroxidation inhibition properties suggest it might — ergothioneine would operate at a different level than the other known nutritional anti-ferroptotic agents. Selenium supports GPX4 synthesis. Riboflavin supports the parallel FSP1 defense pathway. Vitamin D activates NRF2 transcription. Vitamin E traps radicals and inhibits the enzyme that generates ferroptotic death signals. Ergothioneine would chelate the iron catalyst itself and directly inhibit the peroxidation of the ferroptotic substrate — upstream of all of them.

The Case for Acting Now…
Keep reading with a 7-day free trial
Subscribe to NeuroLipid Notebook to keep reading this post and get 7 days of free access to the full post archives.