

Rethinking the HRT ‘Critical Window’: A Membrane-Inflammatory Model of Estrogen Response

For more than two decades, the “critical window hypothesis” has shaped how clinicians think about hormone replacement therapy (HRT) and brain health.
Observational studies suggested that initiating estrogen therapy near menopause might confer neuroprotective benefits, while starting therapy years later could increase dementia risk.
Yet the clinical literature refuses to behave that neatly.
Some women—even some APOE4 carriers—appear to benefit from hormone therapy. Others experience no benefit. A few studies even report worse Alzheimer’s biomarkers with treatment.
Same age.
Same menopausal stage.
Sometimes even the same therapy.
So what explains the difference?
One possibility is that the “window” governing estrogen’s neurological effects is not purely chronological—but biochemical.
Rather than being determined only by years since menopause, the brain may transition through a membrane-inflammatory threshold—a lipid signaling state that determines whether estrogen acts as a stabilizing or destabilizing influence within neural tissue.
If this idea is correct, the critical window-determining the optimal time to begin hormone replacement/optimization therapy may not be a clock.
It may be a lipid threshold.
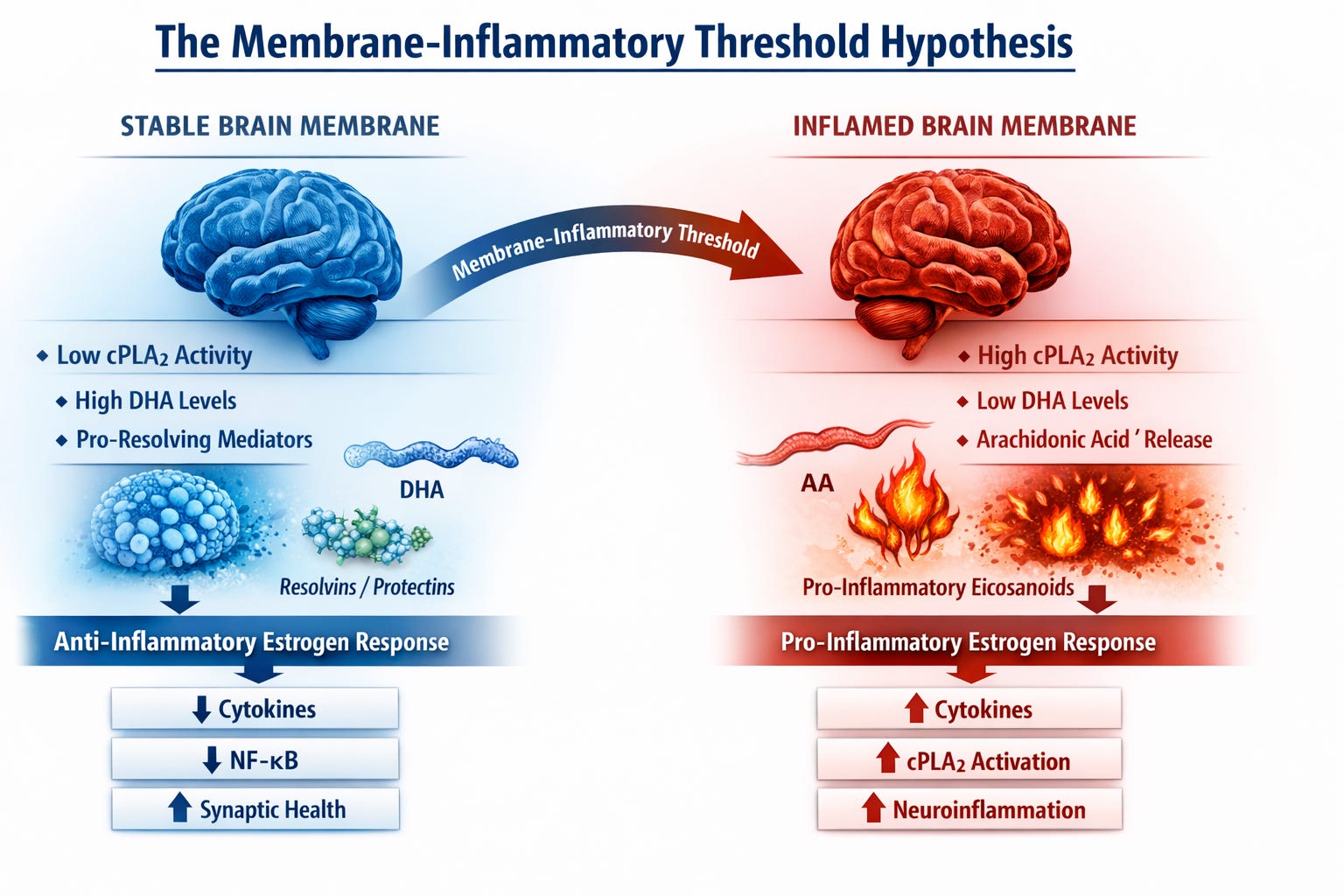
The Brain’s Membrane Lipid Reserve
Neurons are built on membranes.
Every synapse, receptor complex, and signaling cascade operates within a phospholipid bilayer whose composition strongly influences neuronal resilience.
Healthy neuronal membranes are enriched in docosahexaenoic acid (DHA), an omega-3 fatty acid that maintains membrane fluidity and generates specialized pro-resolving mediators that actively terminate inflammation.
These membranes function as a kind of biochemical reserve system.
When lipid composition is stable—characterized by high DHA availability and balanced phospholipid turnover—neurons can absorb metabolic and inflammatory stress without triggering damaging signaling cascades.
But when this reserve erodes, membranes begin to behave differently.
Phospholipid turnover increases. Inflammatory lipid mediators rise. And signaling pathways that were once tightly regulated become chronically activated.
At the center of this transition sits a key enzyme: calcium dependent, cytosolic phospholipase A2 (cPLA2).
cPLA2 and the APOE4 Lipid Inflammatory Axis
One of the most important mechanistic discoveries in APOE4 biology involves the enzyme cPLA2.
In a landmark 2022 study, Hussein Yassine and colleagues established that cPLA2 signaling is markedly hyperactivated in APOE4 carriers.
Across astrocytes, mouse models, and human post-mortem brain tissue they demonstrated:
- increased cPLA2 phosphorylation
- elevated enzymatic activity
- increased production of inflammatory lipid mediators such as leukotriene B4 (LTB4)
cPLA2 functions as a molecular switch in membrane biology.
When activated, it cleaves arachidonic acid (AA) from membrane phospholipids. That arachidonic acid is then converted into pro-inflammatory eicosanoids, amplifying neuroinflammatory signaling.
The result is a shift toward an AA-dominant lipid environment.
Yassine’s lipidomic analyses of human Alzheimer’s brains confirmed widespread activation of the eicosanoid lipidome, with particularly strong signals in APOE4 carriers.
Whether cPLA2 activation is a cause or consequence of neurodegeneration remains debated.
But its role as a central regulator of inflammatory lipid signaling in the APOE4 brain is now well established.
Estrogen and Membrane Biology
Estrogen is widely recognized as neuroprotective and anti-inflammatory.
Through genomic signaling via estrogen receptors—particularly ERβ—estrogen can:
- suppress NF-κB signaling
- reduce pro-inflammatory cytokines
- promote anti-inflammatory microglial states
- enhance synaptic resilience
However, estrogen signaling is not exclusively anti-inflammatory.
Rapid membrane-associated estrogen signaling can activate kinase pathways—including ERK and MAPK—that are known upstream activators of cPLA2.
This creates a fascinating possibility.
The same hormone that suppresses inflammatory signaling through genomic pathways may also activate lipid signaling cascades that liberate arachidonic acid from neuronal membranes.
Which pathway dominates may depend on the baseline lipid state of the membrane itself.
The Membrane-Inflammatory Threshold
This leads to a refinement of the traditional “critical timing window” concept.
Rather than a window defined solely by time since menopause, estrogen’s neurological effects may depend on the lipid signaling state of neuronal membranes.
In a stable membrane environment—characterized by adequate DHA levels and low cPLA2 activity—estrogen signaling may reinforce anti-inflammatory pathways and support synaptic resilience.
But if membranes have already shifted toward an inflammatory lipid state—through DHA depletion and elevated cPLA2 activity—the biochemical context changes.
In that environment, estrogen signaling may increasingly engage rapid kinase pathways that further activate cPLA2, amplifying inflammatory lipid cascades.
Under this model, estrogen’s neurological effects become context-dependent, determined by the inflammatory state of neuronal membranes at the moment therapy begins.
Menopause and the Collapse of Membrane Reserve
Menopause does more than reduce circulating estrogen.
It also alters lipid metabolism and membrane composition.
Estrogen enhances DHA synthesis and incorporation into phospholipids, and women typically exhibit higher DHA concentrations than men during their reproductive years. Hormone therapy increases erythrocyte DHA levels by approximately 11%, while postmenopausal women who are not on estrogen replacement therapy show significantly lower DHA concentrations compared to premenopausal women.
With the loss of estrogen signaling:
- DHA incorporation into membranes declines
- phospholipid remodeling shifts
- inflammatory lipid mediators increase
This suggests that menopause may accelerate the erosion of neuronal membrane reserve.
In this framework, the biological “clock” governing HRT response may reflect not simply time since menopause, but how far membrane lipid stability has deteriorated during that period.
Why the HRT–APOE4 Literature Is So Conflicting
Studies examining hormone therapy in APOE4 carriers have produced strikingly inconsistent results.
Some studies suggest benefit, including analyses from the EPAD cohort, which reported improved cognition and larger brain volumes among APOE4-positive women receiving hormone therapy.
Others report the opposite.
A recent analysis by Jauregi-Zinkunegi and colleagues found that menopausal hormone therapy was associated with worse Alzheimer’s biomarker profiles—specifically CSF p-tau/Aβ42 and Aβ42/40 ratios—in APOE4 carriers. Notably, younger age at MHT initiation (absolute age, not timing relative to menopause) was associated with worse biomarker levels, and this effect was specific to APOE4 carriers.
This finding directly contradicts the classic critical window hypothesis, which predicts that earlier initiation should be protective.
Still others report no genotype-specific effects at all.
If APOE4 carriers vary widely in their baseline membrane lipid state, these contradictions become easier to understand.
Two individuals with identical genetics and menopausal timing may have dramatically different brain lipid environments depending on:
- omega-3 intake
- metabolic health
- inflammatory burden
- lipid transport efficiency
Under this model, the same hormone therapy could produce opposite outcomes depending on whether the brain has crossed a membrane-inflammatory threshold.
DHA and the Stability of Neural Membranes
The balance between arachidonic acid (AA) and docosahexaenoic acid (DHA) plays a central role in membrane signaling.
AA serves as the precursor to many pro-inflammatory eicosanoids.
DHA, by contrast, generates specialized pro-resolving mediators that actively terminate inflammatory responses.
Experimental studies show that DHA suppresses amyloid-β-induced:
- cPLA2 phosphorylation
- nitric oxide production
- TNF-α expression
- oxidative stress
Shifting membranes toward DHA dominance may therefore dampen inflammatory signaling downstream of cPLA2 activation.
However, APOE4 carriers appear to experience accelerated DHA turnover, likely due to increased phospholipid remodeling.
As a result, they may require greater DHA availability to maintain membrane stability.
A Predictive Framework for Alzheimer’s Risk in Women
If membrane lipid stability plays a central role in neuronal resilience, it raises a provocative possibility: the earliest stages of Alzheimer’s risk in women may involve changes in membrane lipid reserve long before classical biomarkers appear.
In this framework, the transition from neurological resilience to vulnerability may unfold in stages.
In the earliest stage, neuronal membranes maintain high DHA availability and balanced phospholipid turnover, allowing synapses to tolerate metabolic and inflammatory stress.
As lipid reserve begins to erode—through declining estrogen signaling, altered lipid transport, or APOE4-associated phospholipid remodeling—membranes shift toward an arachidonic-acid dominant state, increasing activation of enzymes such as cPLA2 and amplifying inflammatory lipid signaling.
Only later do the classical hallmarks of Alzheimer’s disease emerge, including amyloid accumulation, tau pathology, and measurable neurodegeneration.
If this model is correct, the lipid composition of neuronal membranes may function as an early determinant of brain resilience, influencing how neurons respond to both metabolic stress and hormonal signaling.
In this context, the neurological response to hormone therapy may depend not only on chronological timing but on whether the brain’s membrane lipid reserve remains intact.
A New Way to Think About the Critical Window
Alzheimer’s disease research increasingly recognizes that lipid metabolism, inflammation, and neuronal signaling are deeply interconnected.
Within this network, cPLA2 sits at a critical junction, linking membrane composition to inflammatory cascades and neuronal vulnerability.
Yassine’s demonstration of cPLA2 hyperactivation in APOE4 carriers provides a mechanistic foundation for understanding genetic risk.
Extending that insight to hormone therapy suggests a new possibility.
The benefits and risks of estrogen may depend not only on when therapy begins, but on the lipid environment of the brain at the moment estrogen arrives.
If membrane lipid state is modifiable—through dietary, metabolic, or pharmacologic strategies—then the threshold governing estrogen’s effects may itself be a target for intervention.
If this model proves correct, the future of hormone therapy may involve more than timing.
It may involve preserving or restoring the brain’s membrane lipid reserve so that estrogen arrives in a state of stability rather than inflammation.
The critical window, in other words, may not be “within 10 years since final menstrual period”.
It may be a threshold in the biology of neuronal membranes.
But if such a threshold exists, an obvious question follows: can it be measured?
In the upcoming companion article for paid subscribers, I explore several candidate biomarkers that may help approximate the brain’s membrane-inflammatory state—including lipidomic markers, omega-3 indices, and indicators of cPLA₂ pathway activation—and how these measurements could eventually guide precision hormone therapy in APOE4 carriers.
In a subsequent follow-up, I’ll also examine emerging dietary, metabolic, and pharmacologic strategies that may influence membrane lipid biology and could, in theory, help shift the membrane state before hormone therapy begins.
Because if the membrane-inflammatory threshold is real, the future of HRT may not simply be about when estrogen is started.
It may be about whether the brain has been primed to receive it.
If my proposed NeuroLipid Threshold Hypothesis resonates with you, sharing this NeuroLipid Notebook article would be greatly appreciated.