
Rethinking APOE4: cPLA2, Membrane Vulnerability, and Why Fat Loss Drives Brain Inflammation
Why Preserving Neuronal Membranes Matters More Than Simply Adding Omega-3s

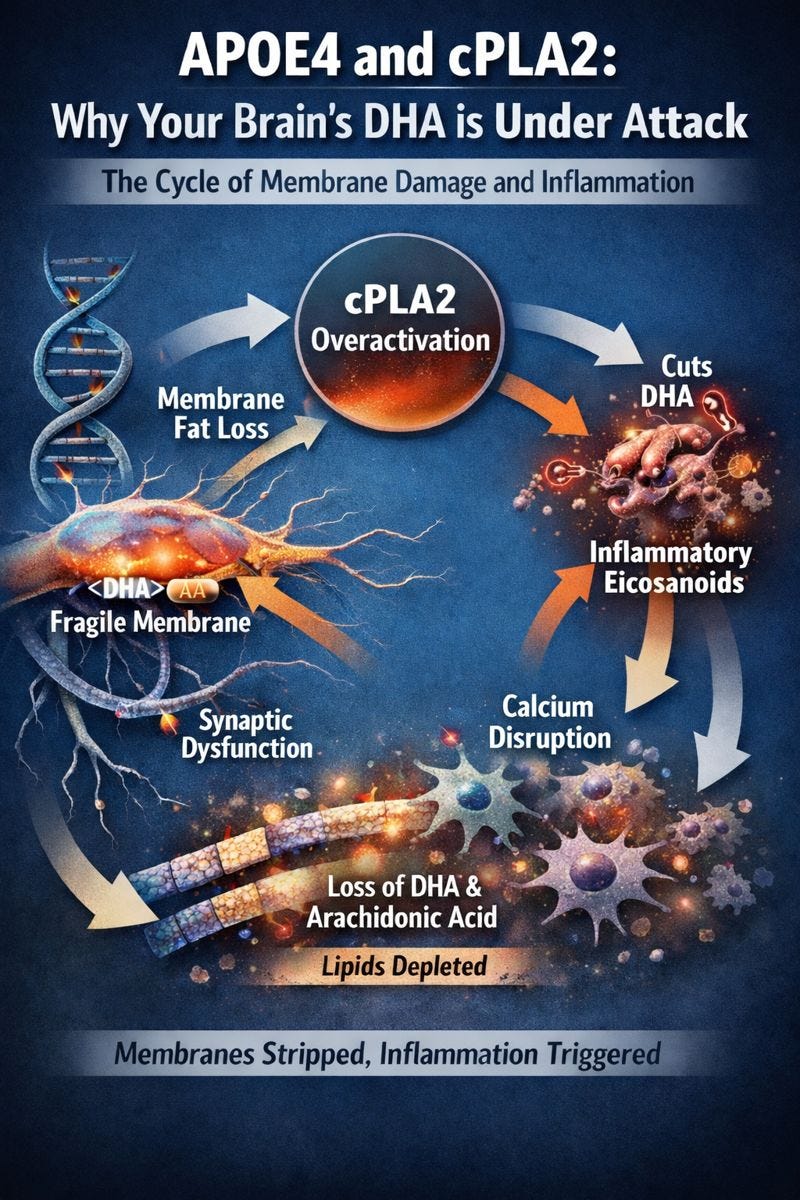
Most people have heard that the APOE4 gene causes Alzheimer’s disease by producing amyloid plaques. If you’re reading this Substack, you probably know it’s way more complicated than that! While it’s partly true, it misses the bigger story: APOE4 fundamentally changes how your brain handles fats. It weakens the protective outer layer of neurons—their membranes—making them fragile and more vulnerable to damage. You can think of it like a house built with thinner walls: even small knocks or stressors can create cracks.
The trouble begins with cPLA2, an enzyme that normally helps brain cells release fatty acids when they’re needed for signaling or to respond to injury. In healthy brains, this is a useful process. In APOE4 carriers, however, cPLA2 is chronically overactive. It keeps cutting fats from membranes even when the brain doesn’t need it. This includes DHA, the omega-3 fat critical for maintaining healthy neuronal membranes, and arachidonic acid, which can trigger inflammation when freed.
APOE4 and cPLA2 form a self-perpetuating vicious cycle. APOE4 disrupts the composition of membrane phospholipids even before cPLA2 activates. Carriers have lower levels of PIP2 due to elevated synaptojanin-1, altered ratios of polyunsaturated phosphatidylethanolamines, and more monounsaturated fatty acids overall. These changes make membranes physically weaker and primed for enzymatic cleavage. When stress or calcium fluctuations trigger cPLA2, the enzyme cleaves DHA and arachidonic acid from the membrane. This not only removes protective fats but also disrupts lipid rafts and receptor scaffolding, impairing the neuron’s ability to communicate and respond.
The problem compounds itself. Membrane damage affects calcium channels and buffering inside the cell, creating unstable calcium signaling, which further activates cPLA2. Meanwhile, arachidonic acid is converted to pro-inflammatory molecules like leukotriene B4, and reactive oxygen species accumulate, damaging the membrane even more. Over time, this creates a feedback loop where membranes are stripped of their protective fats while inflammation escalates. APOE4 behaves as a loss-of-function for membrane maintenance: it fails to deliver lipids efficiently while simultaneously promoting the enzymatic machinery that breaks them down.
This cycle also explains why omega-3 supplementation alone isn’t enough. Even when DHA successfully reaches the brain, overactive cPLA2 continues to remove it from membranes. Lipidomic studies of APOE4 Alzheimer’s brains show low DHA-derived neuroprotectin D1 alongside elevated pro-inflammatory eicosanoids, confirming that supplementation alone cannot overcome the enzymatic stripping. In other words, giving DHA is like filling a bathtub with the drain wide open: some fat gets in, but it leaks out as quickly as it arrives.
That’s why researchers like Hussein Yassine are now thinking of cPLA2 not just as a marker of inflammation, but as the valve controlling membrane integrity. Studies show that reducing or inhibiting cPLA2 protects neurons from Aβ-induced damage, preserves memory, maintains synaptic markers, and prevents additional enzymatic cascades, even when amyloid is present. Targeting cPLA2 directly, rather than just adding fats, interrupts the degenerative loop at its source.
For prevention, this matters most decades before any memory loss appears. Brain-penetrant, selective cPLA2 inhibitors could stabilize membranes, preserve lipid rafts, reduce inflammatory signaling, and allow endogenous or supplemented omega-3s to remain incorporated. By addressing the enzymatic control point, we can shift from “replace what’s lost” to “prevent the loss from happening.” It’s a subtle but critical difference—especially for APOE4 carriers, where early intervention may preserve decades of neuronal health.
In short, Alzheimer’s in APOE4 carriers isn’t just about failing to supply fats—it’s about losing the fats already in place. cPLA2 is the valve that determines whether neuronal membranes are preserved or progressively dismantled. Understanding this gives us a new way to think about prevention: stop the drain, not just fill the tub.
For further reading, please read this monumental post from Hussein Yassine:

As well as his recent scholarly publication on this: https://www.nature.com/articles/s44386-025-00035-0
Don’t Stop Feeding the Brain
APOE4 keeps cPLA2 overactive, continuously stripping DHA and other protective fats from neuronal membranes. It’s a vicious cycle: even the DHA your brain receives is at risk of being lost if we don’t address the enzymatic drain. The literature suggests that APOE4 carriers face a fundamental problem of impaired DHA delivery to the brain—approximately 24% lower brain DHA uptake—due to compromised transport across the blood-brain barrier and defective lipidation.
But that doesn’t mean we give up. Every bit of DHA that reaches the brain helps preserve membrane integrity and supports neuronal health. This is why efficient delivery matters—phospholipid forms like LPC-DHA can cross into the brain more effectively than standard fish oil, giving membranes a better chance to stay resilient.
You can learn more here: https://www.fenixhealthscience.com/jeanniecapone
Disclaimer: I may receive a financial benefit if you choose to purchase through this link.
Feeding the brain DHA is a critical first line of defense. I am really interested in Mr. Yassine’s new company, PebRX, which is striving to determine a candidate therapy to inhibit the overactivation of cPLA2 in fellow APOE4 carriers like myself! Please follow this space , and his research, closely!
This is a really helpful reframing: APOE4 risk isn’t only about plaques, but it’s also about membrane biology! The way you connect overactive cPLA2 to a “leaky” lipid economy (liberating DHA/PUFAs from neuronal membranes, feeding downstream eicosanoid signaling, and sustaining inflammation) makes the story feel much more mechanistically actionable than the usual amyloid-only narrative. 
Two points I especially appreciated:
1. “Just take omega-3” may be too simplistic for APOE4. If APOE4 shifts fatty-acid handling toward higher turnover/oxidation or altered delivery to the CNS, then the bottleneck isn’t necessarily intake, but it’s where the DHA ends up and whether it stays incorporated where it matters (membranes). 
2. The idea (as you outline) that the brain may be pulling in DHA as compensation—not sufficiency—is a subtle but clinically important distinction, and it aligns with how fragile systems behave under chronic stress: higher flux can coexist with lower effective reserves. 
Also: spotlighting Hussein Yassine’s line of work helps anchor this in real translational science, especially the focus on APOE genotype, DHA delivery/uptake, and inflammatory lipid pathways. 
This post makes a strong case that membrane vulnerability + lipid signaling should be treated as a first-class target in APOE4, not an afterthought.